
Choroba Pompego to rzadka choroba spichrzeniowa, w której brak enzymu kwaśnej maltazy prowadzi do odkładania glikogenu w mięśniach, przeponie i sercu. W praktyce oznacza to stopniowe osłabienie siły mięśniowej, problemy z oddychaniem, a u części chorych także objawy kardiologiczne już we wczesnym dzieciństwie. Poniżej porządkuję najważniejsze informacje: od objawów i diagnostyki po leczenie dostępne w Polsce i codzienne funkcjonowanie z tą chorobą.
Najważniejsze informacje o niedoborze kwaśnej maltazy i leczeniu
- To choroba genetyczna dziedziczona autosomalnie recesywnie, związana z mutacjami genu GAA.
- Występują dwie główne postacie: niemowlęca, zwykle przed 1. rokiem życia, oraz późna, która może ujawnić się nawet w dorosłości.
- Najczęstsze sygnały ostrzegawcze to wiotkość mięśni, trudności z oddychaniem, szybkie męczenie się i problemy z wchodzeniem po schodach.
- Rozpoznanie zaczyna się zwykle od badania aktywności enzymu, a potwierdza je diagnostyka genetyczna.
- Podstawą leczenia jest enzymatyczna terapia zastępcza, prowadzona w Polsce w ramach programu lekowego.
- Im wcześniej choroba zostanie rozpoznana i leczona, tym większa szansa na spowolnienie jej postępu i zachowanie sprawności.
Czym jest choroba Pompego i co dzieje się w mięśniach
Najprościej mówiąc, problem polega na tym, że organizm nie rozkłada prawidłowo glikogenu, czyli materiału zapasowego magazynowanego w komórkach. Gdy brakuje enzymu GAA, glikogen gromadzi się tam, gdzie nie powinien, przede wszystkim w mięśniach szkieletowych, mięśniach oddechowych, przeponie i mięśniu sercowym. To właśnie dlatego objawy nie ograniczają się do jednego narządu.
Jest to choroba dziedziczona autosomalnie recesywnie, więc dziecko choruje wtedy, gdy odziedziczy wadliwą kopię genu od obojga rodziców. W praktyce oznacza to też, że choroba może pojawić się w rodzinie nagle, bez wcześniejszej historii rozpoznania. Zdarza się około 1 na 40 000 do 1 na 100 000 urodzeń, choć częstość zależy od populacji.
Wyróżnia się dwie główne postacie. W niemowlęcej objawy pojawiają się przed ukończeniem 1. roku życia i zwykle szybko postępują. W późnej postaci pierwsze sygnały mogą wystąpić po 12. miesiącu życia, a czasem dopiero u dorosłych. Ja zwracam uwagę przede wszystkim na to, że ta sama choroba może wyglądać bardzo różnie, więc brak dramatycznego początku nie wyklucza poważnego problemu.
To ważne także dlatego, że wczesne objawy bywają mylone z „osłabieniem kondycji”, problemem ortopedycznym albo niespecyficzną męczliwością. Następny krok to przyjrzenie się temu, jak choroba objawia się w różnych grupach wiekowych.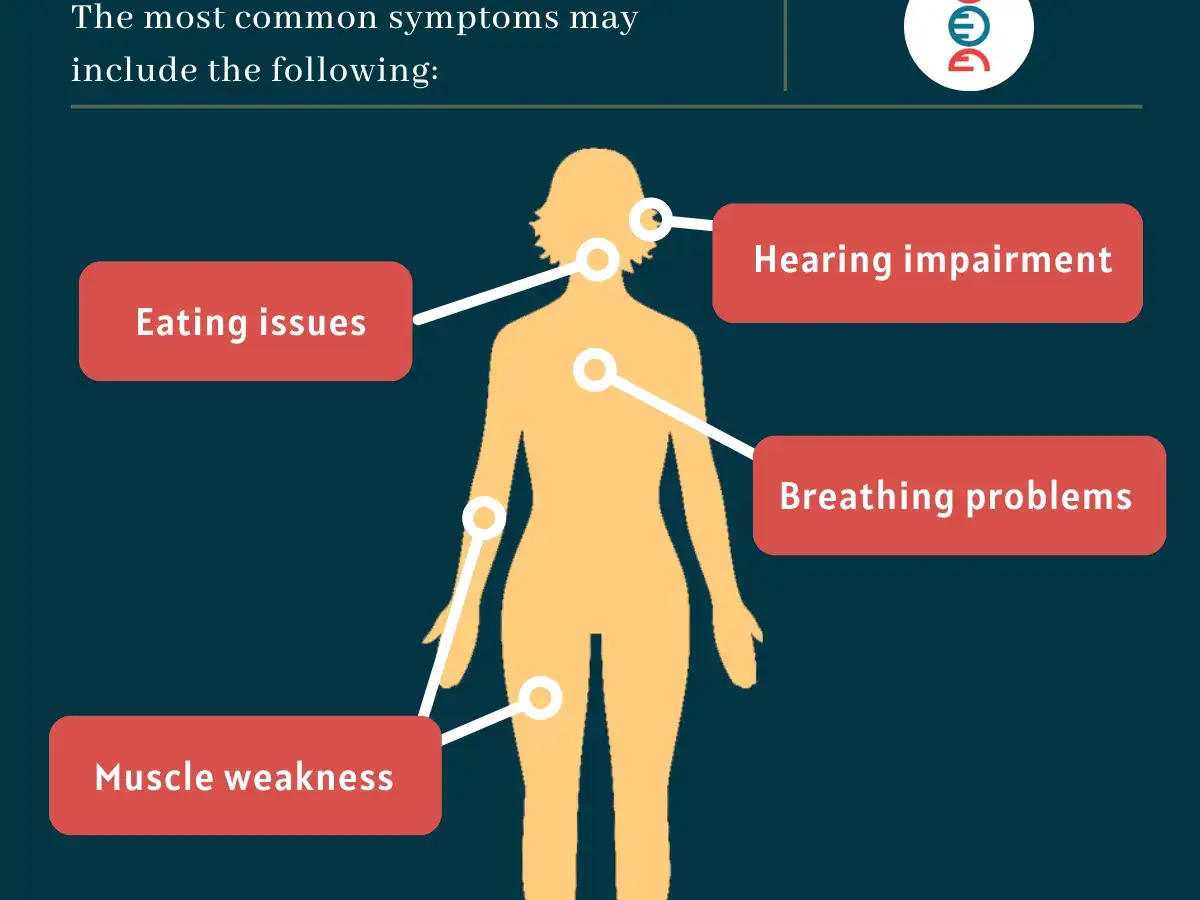
Jakie objawy powinny zwrócić uwagę
Objawy zależą głównie od tego, ile aktywności enzymu zostało zachowanej. Im mniej enzymu, tym cięższy i szybszy przebieg. W postaci niemowlęcej obraz bywa gwałtowny, a w późnej chorobie tempo zmian jest wolniejsze, ale przez to łatwiej ją przeoczyć.
| Początek | Przed 12. miesiącem życia | Po 12. miesiącu życia, czasem dopiero w wieku dorosłym |
|---|---|---|
| Dominujące objawy | Wiotkość mięśni, problemy z karmieniem, słaby przyrost masy ciała, kardiomiopatia, częste infekcje dróg oddechowych | Osłabienie obręczy biodrowej i barkowej, trudności z wstawaniem, schodami i bieganiem, bóle mięśni, poranne bóle głowy |
| Oddech | Szybko pojawiają się trudności z oddychaniem i karmieniem | Często najpierw pojawia się osłabienie mięśni oddechowych, a dopiero później wyraźne trudności ruchowe |
| Serce | Zwykle wyraźnie zajęte | Kardiomiopatia jest rzadsza |
| Ryzyko bez leczenia | Bardzo wysokie, z szybkim pogarszaniem stanu ogólnego | Zmienny przebieg, ale postęp choroby może stopniowo ograniczać samodzielność |
Ważny szczegół, który często umyka: osłabienie oddechu może pojawić się zanim chory zacznie wyraźnie gorzej chodzić. To właśnie dlatego przy przewlekłej duszności, porannych bólach głowy, senności w dzień albo chrapaniu z bezdechami trzeba myśleć szerzej niż tylko o „słabej formie”. Z tego miejsca naturalnie przechodzimy do diagnostyki.
Jak rozpoznaje się tę chorobę
Diagnoza bywa trudna, bo objawy nie są jednoznaczne. W praktyce lekarz zaczyna od rozmowy i badania neurologicznego, ale samo wrażenie kliniczne nie wystarcza. Potrzebne są badania, które pokażą aktywność enzymu i potwierdzą mutacje genu.
Pierwszy krok to zwykle oznaczenie aktywności kwaśnej alfa-glukozydazy w suchej kropli krwi, czyli tzw. DBS. To proste badanie przesiewowe, ale wynik dodatni nie zamyka sprawy. Trzeba go potwierdzić badaniem genetycznym, a czasem także oznaczeniem aktywności enzymu w leukocytach lub fibroblastach.
W diagnostyce często przydają się też badania uzupełniające, bo pomagają ocenić stopień zajęcia narządów i odróżnić tę chorobę od innych przyczyn osłabienia mięśni:
- spirometria w pozycji siedzącej i leżącej,
- ocena siły mięśniowej i funkcji ruchowych,
- badania serca, zwłaszcza echo i EKG,
- badania snu, jeśli pojawiają się bezdechy lub poranne bóle głowy,
- ocena połykania, gdy występują zachłyśnięcia lub problemy z jedzeniem,
- oznaczenie CK, czyli kinazy kreatynowej, która bywa podwyższona w chorobach mięśni.
Ja traktuję tę diagnostykę jak układankę: jeden test mało kiedy wystarcza, ale zestaw wyników bardzo dobrze pokazuje kierunek. Właśnie dlatego przy niejasnym osłabieniu mięśni i niewydolności oddechowej nie warto czekać miesiącami na „samo przejdzie”. Kolejna sekcja pokazuje, co można realnie zrobić po postawieniu rozpoznania.
Jak wygląda leczenie i opieka w Polsce
Podstawą leczenia jest enzymatyczna terapia zastępcza (ERT), czyli podawanie brakującego enzymu dożylnie. W Polsce takie leczenie prowadzi się w ramach programu lekowego, a infuzje odbywają się zwykle co 2 tygodnie w warunkach szpitalnych. To nie jest drobna interwencja, ale dla wielu chorych właśnie ona zmienia tempo choroby.
Najważniejsza zasada brzmi: im wcześniej terapia zostanie rozpoczęta, tym większa szansa na spowolnienie uszkodzeń mięśni i oddechu. U niemowląt dodatkowe znaczenie ma też kwestia odpowiedzi immunologicznej na podawany enzym, dlatego lekarze czasem oceniają tzw. status CRIM, czyli to, czy organizm wytwarza choć część własnego białka i jak może reagować na leczenie.
Sama infuzja nie wyczerpuje jednak leczenia. Dobra opieka obejmuje kilka równoległych elementów:
- regularną fizjoterapię dopasowaną do wydolności pacjenta,
- kontrole pulmonologiczne i monitorowanie funkcji oddechowej,
- opiekę kardiologiczną, szczególnie w postaci niemowlęcej,
- wsparcie żywieniowe, jeśli pojawiają się trudności z karmieniem lub połykaniem,
- szczepienia ochronne i ograniczanie infekcji, które mogą szybko pogorszyć stan oddechowy.
To ważne, bo sama enzymatyczna terapia zastępcza nie naprawia wszystkiego. Najlepsze wyniki daje połączenie leczenia przyczynowego z rehabilitacją, kontrolą oddychania i dobrym prowadzeniem żywieniowym. Z tego wynika też codzienna strategia, o której często mówi się za mało.
Jak żyć z chorobą na co dzień i nie przegapić pogorszenia
W tej chorobie nie opłaca się działać „na wyczucie”. Lepsze efekty daje spokojny, uporządkowany plan opieki niż próby nadrabiania objawów po fakcie. Ja zwykle zachęcam, by myśleć o trzech filarach: ruch, oddech i odżywianie.
W praktyce pomaga:
- regularna rehabilitacja prowadzona przez osoby, które znają choroby nerwowo-mięśniowe,
- obserwacja tolerancji wysiłku, zwłaszcza schodów, wstawania z krzesła i marszu na dłuższym dystansie,
- kontrola snu, bo poranne bóle głowy i senność w ciągu dnia często sugerują nocne problemy oddechowe,
- dbanie o szybkie leczenie infekcji dróg oddechowych,
- konsultacja dietetyczna, gdy pojawiają się trudności z połykaniem lub spadek masy ciała,
- poradnictwo genetyczne dla rodziny, bo to ma znaczenie przy kolejnych ciążach i badaniu rodzeństwa.
Warto też uważać na pułapkę, którą widzę często: ktoś poprawia kondycję „na własną rękę”, forsuje się mimo narastającej duszności, a później trafia do lekarza dopiero wtedy, gdy ograniczenia są już duże. Przy tej chorobie trening bez nadzoru nie jest neutralny. Powinien być dopasowany do wydolności mięśni i oddechu, a nie do ambicji.
Jeśli objawy nasilają się powoli, łatwo je zbagatelizować. Dlatego poniżej wyraźnie oddzielam sygnały, które jeszcze można omówić na planowej wizycie, od takich, przy których nie warto czekać.
Kiedy nie czekać i szybko szukać pomocy
Są sytuacje, w których trzeba działać szybciej niż „przy najbliższej kontroli”. U niemowlęcia niepokój powinny wzbudzić: wyraźna wiotkość, słabe ssanie, brak przyrostu masy ciała, duszność, zasinienie, częste zachłystywanie albo powiększone serce wykryte w badaniach. U starszego dziecka i dorosłego alarmem są narastająca duszność, trudność z oddychaniem na leżąco, częste upadki, pogarszająca się mowa i połykanie oraz szybki spadek sprawności.
Do pilnej konsultacji skłaniają też objawy nocne: bezdechy, budzenie się z bólem głowy, uczucie niewyspania mimo długiego snu i senność w dzień. To nie są drobiazgi. W schorzeniach mięśni oddechowych często właśnie noc mówi najwięcej o tym, co dzieje się w organizmie w ciągu doby.
Jeśli choroba jest choćby podejrzewana, najlepiej trafić do ośrodka, który ma doświadczenie w chorobach nerwowo-mięśniowych. Tam szybciej da się uporządkować badania, ocenić oddech i zdecydować, czy potrzebne jest leczenie wspomagające. A na koniec zostaje rzecz bardzo praktyczna: jak przygotować się do takiej wizyty, żeby nie stracić czasu.
Co przygotować przed wizytą, żeby przyspieszyć diagnostykę
Najbardziej pomaga krótka, ale konkretna dokumentacja. Warto spisać, od kiedy pojawiły się objawy, co się zmieniało i co najbardziej ogranicza codzienne funkcjonowanie. Dobrze działa też prosta oś czasu: pierwsze problemy z chodzeniem, schodami, oddychaniem, snem, karmieniem albo częstymi infekcjami.
- zapis objawów z datami lub przybliżonym wiekiem ich początku,
- wyniki wcześniejszych badań, zwłaszcza CK, spirometrii, EKG i echo serca,
- informacja o chorobach mięśni, niewyjaśnionych zgonach lub podobnych objawach w rodzinie,
- lista leków i suplementów,
- krótki opis tego, co najbardziej pogarsza stan: infekcja, wysiłek, brak snu, schody, leżenie na płasko.
Jeśli lekarz podejrzewa tę chorobę, dobrze od razu zapytać o badanie aktywności GAA i dalsze potwierdzenie genetyczne, zamiast czekać na kolejne, mniej celowane testy. Właśnie taka precyzja na początku często oszczędza miesiące błądzenia. I to jest chyba najważniejsza myśl, z jaką warto zostać: w chorobach rzadkich nie wygrywa ten, kto robi najwięcej badań, tylko ten, kto robi właściwe badania we właściwym momencie.